
Publicatiedatum: 10-10-2022
Inhoudsopgave
2-De noodzaak van de omzetting naar een standaard vergunning wordt niet aangetoond.
3-Alleen studies die een goede werking van de vaccins aantonen lijken van belang.
4-Met een onjuiste aannames effent de EMA de weg naar een standaardvergunning
5-Wat rechtvaardigt dat ook de komende aangepaste vaccins zijn goedgekeurd?
6-Voor wie doet de EMA dit? De patiënten!!!
7-Samenvatting - Tot slot - Ter overdenking
1-Inleiding- het bericht

Op 16 september 2022 heeft de EMA het nieuws naar buiten gebracht dat het aanbeveelt bij de Europese Commissie (EC) om de voorlopige goedkeuring van de COVID-19 vaccins Comirnaty (van Pfizer) en Spikevax (Moderna) om te zetten in een standaard goedkeuring om deze vaccins in Europa te mogen verhandelen. [1]
Hier volgen enkele kritische opmerkingen met betrekking tot de onderbouwing van die aanbeveling in dit nieuwsbericht.
De aangehaalde Engelse citaten zijn ontleend aan het EMA nieuwsbericht van 16-09-2022.
[1] EMA bericht 16-09-2022
https://www.ema.europa.eu/en/news/ema-recommends-standard-marketing-authorisations-comirnaty-spikevax-covid-19-vaccines
2-De noodzaak van de omzetting naar een standaard vergunning wordt niet aangetoond.
Citaat
…” These no longer need to be renewed annually. All other obligations for the companies remain in place.”…
Het is nu niet meer nodig om deze (voorwaardelijke vergunningen) jaarlijks opnieuw te verlengen. Verder blijven alle andere verplichtingen voor de bedrijven van kracht.
Toelichting
Gesuggereerd wordt dat het slechts gaat om administratieve handeling. Er verandert immers volgens de EMA verder niets. De verplichtingen blijven bestaan. In een voetnoot worden de voorwaarden genoemd waaronder het was toegestaan om de COVID-19 vaccins Comirnaty en Spikevax in handel te brengen.
i) een positieve baten-risicobalans,
ii) het feit dat de vaccins moesten worden gebruikt in het kader van een noodsituatie op het gebied van de volksgezondheid,
iii) de waarschijnlijkheid dat de aanvragers na de toelating uitgebreidere klinische en kwaliteitsgegevens zouden kunnen verstrekken.
Maar als er niets verandert wat is dan de noodzaak om de omzetting van een voorlopige in een standaard vergunning door te voeren?
Analyse
Op 16 september 2022 wordt aan de EC voorgesteld de voorlopige vergunning voor het verhandelen van de COVID-19 vaccins Comirnaty (Pfizer) en Spikevax (Moderna) om te zetten in een gewone standaard vergunning. Dit is opmerkelijk omdat de fase 3 onderzoeken van Pfizer en Moderna nog niet zijn afgerond. Bijzonder is ook dat de omzetting naar een standaard vergunning tegelijkertijd geldt voor de nieuwe en de toekomstige vaccin varianten
In de twee weken die hieraan vooraf gingen, zijn nieuwe bivalente boostervaccins goedgekeurd. Deze vaccins beschermen niet alleen tegen het originele virus maar ook tegen verschillende Omicron varianten.
Zo zijn op 1 september 2022 twee bivalente boosters voor goedkeuring aan de EC voorgelegd. Het gaat om het Original Comirnaty/Omicron BA.1 vaccin van Pfizer en het Original Spikevax/Omicron BA.1 vaccin van Moderna [2]. Daarna wordt in het persbericht van 12 september 2022 melding gemaakt dat nog een booster van Pfizer voor goedkeuring aan de EC is voorgelegd: het Comirnaty Original / Omicron BA.4-5 boostervaccin. Opmerkelijk detail is dat in het persbericht staat dat de klinische onderzoeken nog niet zijn afgerond. [3]

Met deze nog niet afgeronde onderzoeken lijkt een administratieve omzetting daarom niet logisch. De EMA denkt daar uiteraard anders over.
[2] EMA bericht 01-09-2022:
https://www.ema.europa.eu/en/news/first-adapted-covid-19-booster-vaccines-recommended-approval-eu
[3] EMA bericht 12-09-2022:
https://www.ema.europa.eu/en/news/adapted-vaccine-targeting-ba4-ba5-omicron-variants-original-sars-cov-2-recommended-approval
3-Alleen studies die een goede werking van de vaccins aantonen lijken van belang.
Citaat
…”These trials and additional studies, including observational studies, have provided reassuring data on key aspects such as how well the vaccines prevent severe COVID-19. In addition, the companies have provided all requested additional data on the pharmaceutical quality of the vaccines.”…
Deze onderzoeken en aanvullende studies, waaronder observationele studies, hebben geruststellende gegevens opgeleverd over belangrijke aspecten, zoals hoe goed de vaccins ernstige gevolgen van COVID-19 voorkomen. Daarnaast hebben de bedrijven alle gevraagde aanvullende gegevens over de farmaceutische kwaliteit van de vaccins verstrekt.
Analyse
Volgens het bericht zet de EMA deze stap omdat diverse studies positieve resultaten laten zien over de preventieve werking van de vaccins. Om welke studies het gaat wordt niet vermeld. Het is toch een kleine moeite om met een link daar naar te verwijzen. Maar een glimp van die geruststellende data wordt ons hier niet gegund. Ook geen woord over de ernstige bijwerkingen en overlijdensgevallen die zijn geregistreerd bij de EMA in EudraVigilance. Verder wordt er expliciet op gewezen dat aan de formele verplichting is voldaan door het leveren van aanvullende data. Er is geen enkele verwijzing naar de inhoud, de kwaliteit, de omvang en de relevantie van die data. We weten dus niet tot welke inzichten dat heeft geleid anders waren daar zeker wel enkele woorden aan gewijd. Het hebben voldaan aan de verplichting lijkt van groter belang dan de inhoud waarmee aan die verplichting is voldaan.
4-Met onjuiste aannames effent de EMA de weg naar een standaardvergunning
Citaat:
…” Taking into account the totality of the available efficacy and safety data resulting from the large utilisation of these vaccines, the specific obligations are no longer considered key to the benefit-risk (of the products), which has cleared the way to move from a conditional to a standard marketing authorisation.”…
Het omvangrijke gebruik van deze vaccins heeft een totaal van de beschikbare gegevens over werkzaamheid en veiligheid opgeleverd.
Dit betekent dat het niet langer nodig is om dit als een specifieke verplichting te handhaven die essentieel is voor de baten-risico analyse (van de producten).
Hierdoor is de weg vrijgemaakt om van een voorwaardelijke naar een standaardvergunning voor het in de handel brengen over te gaan.
De kern van de kettingredenering (A -> B -> C)
Aanname 1:
Als we veel gegevens hebben (A)
dan is dit geen sleutelvoorwaarde meer (B).
Aanname 2:
Als dit geen sleutelvoorwaarde meer is (B)
dan is de weg vrij voor omzetting (C).
Waarom is aanname 1 niet geldig?
De EMA stelt vast dat nu die gegevens er zijn, de beschikbaarheid van data geen essentieel element meer is voor het onderdeel van de baten-lasten van de producten. Die conclusie kan je niet trekken op basis van de beschikbaarheid van gegevens alleen. De gegevens moeten ook juist, betrouwbaar, volledig en actueel zijn.
Dat veel data beschikbaar zijn betekent ook niet automatisch dat aan de sleutelvoorwaarde is voldaan. Met die data moet eerst nog worden aangetoond dat er sprake is van een positieve baten-lasten balans. Als dat al is aangetoond dan zou een link naar de onderliggende documentatie op zijn plaats zijn geweest. Het is waarschijnlijker dat de EMA van mening is dat er een positieve baten-lasten balans is. In dat geval staat A ( de gegevens) los van B (sleutelvoorwaarde)
Alles bij elkaar betekent dit dat de eerste aanname als niet geldig moet worden beoordeeld.
Waarom is aanname 2 niet geldig?
In het begin zijn meerdere voorwaarden opgenoemd die een rol hebben gespeeld bij de voorlopige goedkeuring om de vaccins op de Europese markt te mogen brengen.
De EMA gaat ervan uit dat als aan één van de voorwaarden wordt voldaan alle andere voorwaarden kunnen vervallen. Dit staat nergens. Dit betekent dat ook de tweede aanname niet geldig is.
De conclusie is niet geldig -maar het gaat zo wel gebeuren.
De inhoudelijke grondslag bij de redenering ontbreekt om de voorwaardelijke handelsvergunning van de COVID-19 vaccins van Pfizer en Modena om te zetten in een standaard vergunning. Deze omzetting is kennelijk wenselijk. Dan is alleen de redenering voldoende om deze omzetting er door te drukken
5-Wat rechtvaardigt dat ook de komende aangepaste vaccins zijn goedgekeurd?
Citaat
…” Conditional marketing authorisations are reviewed annually. The CHMP recommended their conversion to standard marketing authorisations as an outcome of the second annual renewal procedure. This recommendation covers all existing and upcoming adapted Comirnaty and Spikevax vaccines, including the recently-approved adapted Comirnaty Original/Omicron BA.1, Comirnaty Original/Omicron BA.4/5 and Spikevax bivalent Original/Omicron BA.1.”…
Voorwaardelijke vergunningen voor het in de handel brengen worden jaarlijks herzien. Het CHMP heeft aanbevolen deze om te zetten in standaardvergunningen voor het in de handel brengen als resultaat van de tweede jaarlijkse verlengingsprocedure. Deze aanbeveling heeft betrekking op alle bestaande en komende aangepaste Comirnaty- en Spikevax-vaccins, inclusief de onlangs goedgekeurde aangepaste Comirnaty Original / Omicron BA.1, Comirnaty Original / Omicron BA.4 / 5 en Spikevax bivalent Original / Omicron BA.1.
Toelichting: EC/EMA/CHMP/CBG
De EMA houdt toezicht op de ontwikkeling, evaluatie en veiligheid van geneesmiddelen inclusief vaccins. De EMA wordt daarbij ondersteund door de CHMP (Committee on Human Medicinal Products') dat nu Committee for Medicinal Products for Human Use wordt genoemd. Nederland wordt daarin vertegenwoordigd door het CBG (Centraal Bureau Geneesmiddelen). De Nederlandse regering is vertegenwoordigd in de Europese Commissie (EC). De EC bekrachtigt de voorstellen van de EMA.
Analyse
Het moment van de tweede verlenging wordt aangegrepen om de verlengingsprocedure in een standaard procedure om te zetten. Zoals hiervoor al is betoogd is er geen inhoudelijke grondslag om de voorwaardelijke verlenging om te zetten in een standard vergunning. De noodzaak om tot een administratieve lastenverlichting te komen wordt niet als argument genoemd. Hiermee lijkt ook de administratieve noodzaak te ontbreken.
De aanbeveling van de EMA heeft betrekking op alle bestaande en aankomende aangepaste Comirnaty- en Spikevax-vaccins. De onderbouwing, waarom dat ook op de nieuwe vaccin varianten van toepassing is, ontbreekt. Er is ook geen link naar de onderliggende documentatie.
Dit besluit zou wel eens onderdeel van een deal kunnen zijn tussen de EU/EMA en de farmaceuten Moderna en Pfizer. Voor het ontwikkelen van nieuwe varianten willen deze farmaceuten markt- en afzetzekerheid.
Door op de voorhand verzekerd te zijn van goedkeuring wordt het ook minder noodzakelijk om uitvoerig te testen. Dat is een kostenbesparing voor de farmaceuten. Of het sneller kunnen beschikken over nieuwe vaccins ten koste gaat van de veiligheid lijkt van latere zorg te zijn.
6-Voor wie doet de EMA dit? De patiënten!!!
Citaat
…” As for any medicine, Comirnaty and Spikevax continue to be closely monitored. EMA will continue to assess any new data promptly and take action to protect patients as needed.”…
Comirnaty en Spikevax zullen net als elk geneesmiddel nauwlettend in de gaten worden gehouden. De EMA zal alle nieuwe gegevens direct blijven beoordelen en als daar aanleiding toe is zal de EMA actie ondernemen om de patiënten te beschermen.
Analyse
We lezen dat de vaccins in de gaten worden gehouden. Maar dat gebeurde al.
Verder staat er dat als nieuwe data daar aanleiding toe geven de EMA direct actie zal ondernemen om de patiënt te beschermen. Hiermee is het oordeel geveld over de (ernstige) bijwerkingen en overlijdensgevallen die zijn geregistreerd bij de EMA in EudraVigilance. Sommigen zouden misschien verwachten dat deze meldingen een rol moeten spelen bij de discussie of deze vaccins uit de markt moeten worden gehaald. Dat is dan een tegenvaller. De meldingen spelen niet eens een rol bij de omzetting naar standaardvoorwaarden. De huidige omvang van de geregistreerde meldingen van bijwerkingen in EudraVigilance is kennelijk acceptabel voor de EC, de EMA en de CHMP. Dus ook voor Nederland die in die organisaties is vertegenwoordigd.
Onduidelijk is waarom de EMA zich bij de bescherming alleen op patiënten richt. Bij vaccinatie gaat het om de preventie van gezonde mensen tegen een ziekte. Dat zijn geen patiënten. Wie de EMA met patiënten voor ogen heeft is onduidelijk. Het lijkt op beroepsdeformatie. Europa als een hospitaal en de burgers als de afhankelijke patiënten waarbij de experts de behandelmethode bepalen.
7- Samenvatting - tot slot – ter overdenking
Onderzoeken lopen nog
Het is opmerkelijk dat de standaardvoorwaarden voor de nieuwe en de toekomstige vaccins varianten gelden. Het lijkt erop dat klinische onderzoeken bij deze en nieuwe vaccins steeds minder belangrijk worden. Bij het oorspronkelijke vaccin van Pfizer werd bij bijna 20.000 personen het vaccin toegediend. De BA.1 variant is getest bij een paar honderd personen. En bij de BA.4-5 varianten waren de klinische onderzoeken niet eens afgerond.
Wel samenwerking met Farmaceuten – geen aandacht vaccinatieschade
De redenering om over te gaan naar standaardvoorwaarden mist een (transparante) inhoudelijke grondslag. Het heeft meer weg van een gelegenheidsargumentatie om de omzetting naar een standaard vergunning gerealiseerd te krijgen.
Het lijkt op samenwerking. Jullie (farmaceuten) blijven varianten ontwikkelen in ruil voor marktzekerheid. De communicatieafdelingen moeten dat resultaat dan verpakken in termen van gezondheidswinst zonder al te veel aandacht te schenken aan gezondheidsschade.
Een positieve baten-lasten balans op commando
De EMA zegt daarover dat als nieuwe gegevens daar aanleiding toe geven actie zal worden ondernomen. Daarmee wordt ook aangegeven dat het huidige niveau van geregistreerde (ernstige) bijwerkingen en overlijdensgevallen in EudraVigilance dus acceptabel is. De EMA stelt gewoon dat er een positieve baten-lasten balans is, terwijl er geen onderzoek is gedaan naar onderrapportage van de meldingen. Bij het vaststellen van een coronadode maakt het niet uit op de persoon met of aan corona is overleden. De 162 officiële overlijdensverklaringen met vaccinatie als oorzaak van overlijden worden minutieus uitgeplozen op aspecten die dit in twijfel kunnen trekken. [4] Bovendien heeft de EMA bij het bepalen van de baten-lasten balans niet vooraf de spelregels en de normen vastgelegd. Dat betekent dat de EMA bij elke uitkomst kan zeggen dat er een positieve baten-lasten balans is.
[4] zie H7
https://www.cbs.nl/nl-nl/longread/rapportages/2022/sterfte-en-oversterfte-in-2020-en-2021
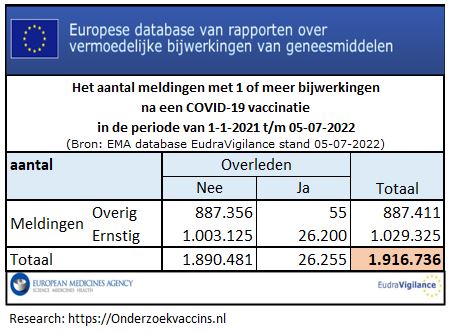
Wel aandacht voor omgangsvormen in de Tweede Kamer maar niet voor de bijna 2 mln geregistreerde bijwerkingen in EudraVigilance (stand 05-07-2022)
Bij dit alles is de Nederlandse regering mede verantwoordelijk voor die besluitvorming na het instemmen van de EC met het omzetten van de voorlopige vergunning in een standaard vergunning. De regering loopt te hoop als de omgangsnormen in de Tweede Kamer in het geding zijn. Gaat het om meer dan 26.000 gemelde overlijdens gevallen en meer dan 1 miljoen ernstige bijwerken, die zijn geregistreerd in EudraVigilance (stand 05-07-2022), dan wordt dat als acceptabel beschouwd.
* * * * *
Naar: Homepage Onderzoekvaccins
Naar: Facebook Onderzoekvaccins
Naar: MeWe Onderzoekvaccins:
Naar: Telegramkanaal Onderzoekvaccins
Abonneer je op het Telegramkanaal en je wordt op de hoogte gehouden van nieuwe publicaties op deze website.
(Abonneren is hetzelfde als volgen op Facebook.)